

敏泰元生物 — 细胞与免疫学专业服务平台
ChIP技术的应用
发布日期:2012-12-10 浏览次数:5867
ChIP技术的应用
将ChIP与第二代测序技术相结合的ChIP-Seq 技术,能够高效地在全基因组范围内检测与组蛋白、转录因子等互作的DNA 区段。ChIP-Seq 的原理是:首先通过染色质免疫共沉淀技术(ChIP)特异性地富集目的蛋白结合的DNA 片段,并对其进行纯化与文库构建;然后对富集得到的DNA 片段进行高通量测序。研究人员通过将获得的数百万条序列标签精确定位到基因组上,从而获得全基因组范围内与组蛋白、转录因子等互作的DNA 区段信息。
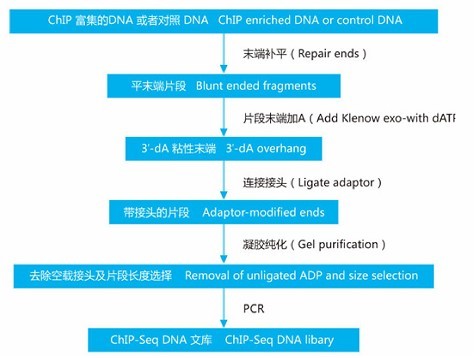 |
| 实验流程示意图 |
 |
| 生物信息分析流程示意图 |
- 1.测序
- 对客户提供的ChIP 样品(如果有阴阳参启动子区域或DNA 序列的)进行定量检测,检测合格后进行测序文库构建、DNA 成簇(Cluster generation)扩增、高通量测序。
- 2.基本数据分析
- 数据产出统计:对测序结果进行图像识别(Base calling),去除污染及接头序列;统计结果包括:测定的序列(Reads)长度、Reads 数量、数据产量。
- 3.高级数据分析
- 标准高级数据分析内容包括:
- (1)ChIP-Seq 序列与参考序列比对;
- (2)Peak calling:统计样品Peak 信息(峰检测及计数、平均峰长度、峰长中位数);
- (3)统计样品Uniquely mapped reads 在基因上、基因间区的分布情况及覆盖深度;
- (4)给出每个样品Peak 关联基因列表及GO 功能注释;
- (5)在多个样品间,对与Peak 关联基因做差异分析。
- 由于ChIP-Seq 的数据是DNA 测序的结果,为研究者提供了进一步深度挖掘生物信息的资源,研究者可以在以下几方面展开研究:
- (1)判断DNA 链的某一特定位置会出现何种组蛋白修饰;
- (2)检测RNA polymerase II 及其它反式因子在基因组上结合位点的精确定位;
- (3)研究组蛋白共价修饰与基因表达的关系;
- (4)CTCF 转录因子研究。
目前,随着人类基因组测序工作的基本完成,研究目的蛋白和整个基因组的相互作用逐渐成为研究的热点。由于基因组中的信息量非常大,常规方法通常无法满足科研的需要。近年来发展起来的ChIP-chip技术将基因组DNA芯片(chip)技术与染色质免疫沉淀技术(ChIP)相结合,为研究目的蛋白与整个基因组相互作用提供了可能。ChIP-chip 技术通过标记染色质免疫沉淀富集的DNA片段,和另一个被标记不同探针的对照组样品一起,与DNA芯片杂交,再利用各种生物信息学方法对收集到的信号进行分析,具体的实验步骤请参考Dr. Richard Young在Nature Protocols上的文章[1]。ChIP-chip技术已经被广泛应用于研究转录因子在整个基因组中的信号网络[2,3,4],染色质修饰机制在基因组中的调控[5, 6],DNA的复制,修复以及修饰[7, 8],基因的转录与核运输[9, 10, 11]等诸多方面。
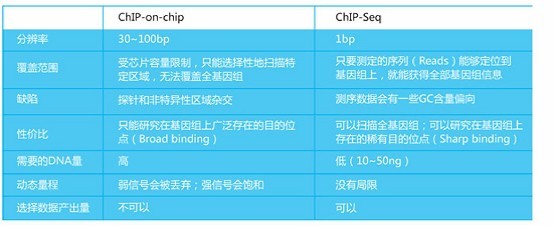 |
| Chip-Chip与Chip-Seq的比较 |
染色质免疫沉淀技术还可用于分析两种蛋白共同结合的DNA序列,即ChIP reChIP方法。ChIP reChIP是在第一次ChIP的基础上不解交联,而继续进行另一个目的蛋白的免疫沉淀,从而得到与两种目的蛋白都结合的DNA序列。值得注意的是,因为通过两次免疫沉淀富集的DNA量比较少,所以在分析时通常要把多次免疫沉淀的DNA浓缩后再进行操作。
- 1.Lee T. I., Johnstone S.E., Young R.A., Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat. Protocols, 1, 729-747, 2006.
- 2.Nal B., Mohr E., Ferrier P.. Location analysis of DNA-bound proteins at the whole-genome level: untangling transcriptional regulatory networks. Bioessays 23, 473-476, 2001.
- 3.Hanlon S.E., Lieb J.D.. Progress and challenges in profiling the dynamics of chromatin and transcription factor binding with DNA microarrays. Curr. Opin. Genet. Dev. 14, 697-705, 2004.
- 4.Banerjee N., Zhang M.Q.. Identifying cooperativity among transcription factors controlling the cell cycle in yeast. Nucleic Acids Res., 31; 7024-7031, 2003.
- 5.Carter N.P., Vetrie D.. Applications of genomic microarrays to explore human chromosome structure and function. Hum. Mol. Genet. 13 (Spec No 2), R297-R302, 2004.
- 6.Van Steensel B., Henikoff S.. Epigenetic profiling using microarrays. Biotechniques, 35, 346-357, 2003.
- 7.MacAlpine D.M., Bell S.P.. A genomic view of eukaryotic DNA replication. Chromosome Res., 13, 309-326, 2005.
- 8.Woodfine K., Carter N.P., Dunham I., Fiegler H.. Investigating chromosome organization with genomic microarrays. Chromosome Res., 13, 249-257, 2005.
- 9.Lei E.P., Krebber H., Silver P.A.. Messenger RNAs are recruited for nuclear export during transcription. Genes. Dev., 15, 1771-1782, 2001.
- 10.Ishii K., Arib G., Lin C., Van Houwe G., Laemmi U.K.. Chromatin boundaries in budding yeast: the nuclear pore connection. Cell, 109, 551-562, 2002.
- 11.Rodriguez-Navarro S. et al. Sus1, a functional component of the SAGA histone acetylase complex and the nuclear pore-associated mRNA export machinery. Cell, 116, 75-86, 2004.
【上一篇】ChIP技术总结
【下一篇】BD PMG协助发现调控T细胞分化增殖的关键因素